This function takes an edgelist and recodes (relabels) the nodes following ape's coding convention.
# S3 method for class 'matrix'
as.phylo(x, edge.length = NULL, root.edge = NULL, ...)
# S3 method for class 'aphylo'
as.phylo(x, ...)Arguments
- x
Either an edgelist or an object of class aphylo.
- edge.length
A vector with branch lengths (optional).
- root.edge
A numeric scalar with the length for the root node (optional).
- ...
Further arguments passed to the method.
Value
An integer matrix of the same dimmension as edges with the following
aditional attribute:
- labels
Named integer vector of size
n. Original labels of the edgelist where the firstnare leaf nodes,n+1is the root node, and the reminder are the internal nodes.
Examples
# A simple example ----------------------------------------------------------
# This tree has a coding different from ape's
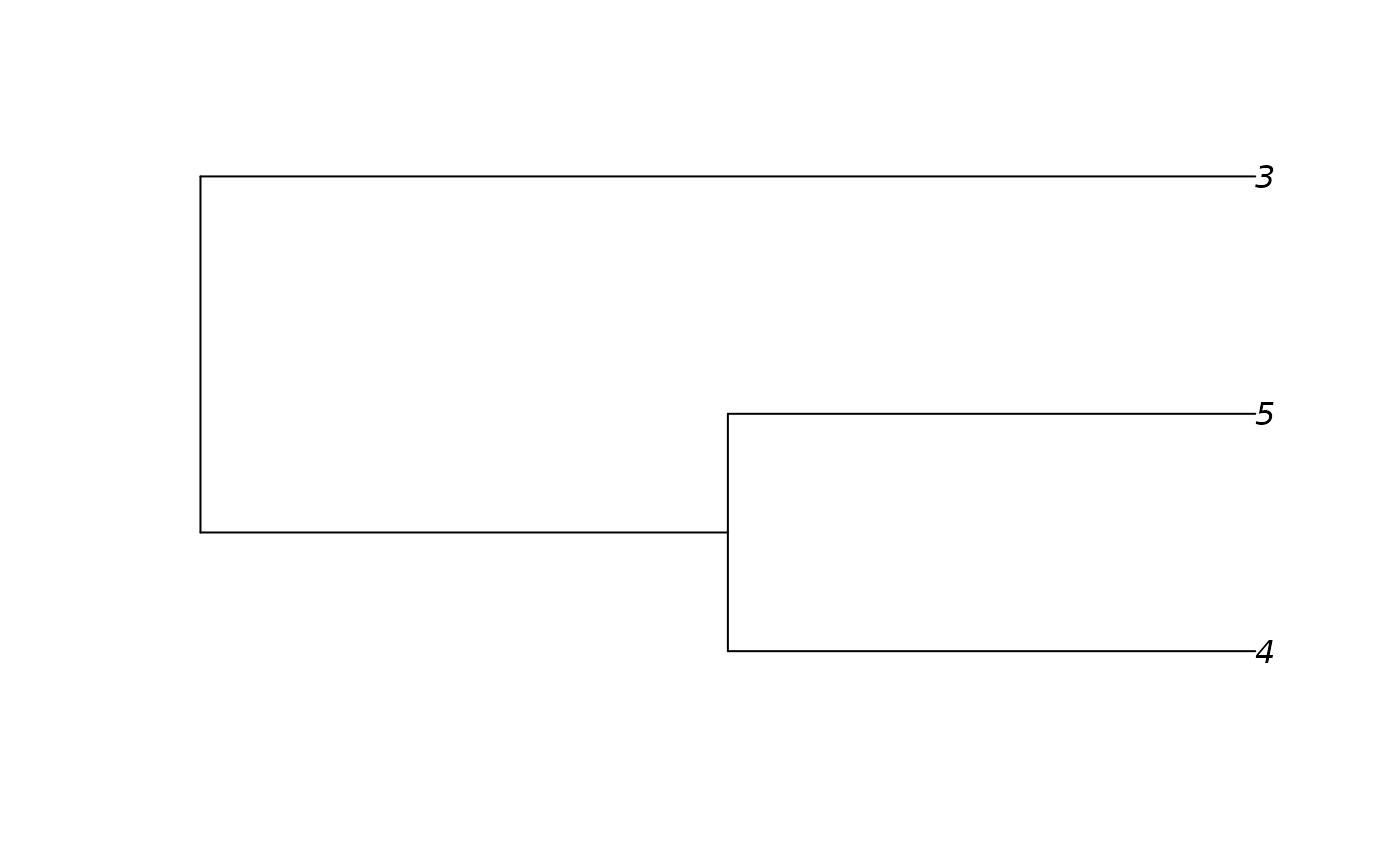
mytree <- matrix(c(1, 2, 1, 3, 2, 4, 2, 5), byrow = TRUE, ncol=2)
mytree
#> [,1] [,2]
#> [1,] 1 2
#> [2,] 1 3
#> [3,] 2 4
#> [4,] 2 5
ans <- as.phylo(mytree)
ans
#>
#> Phylogenetic tree with 3 tips and 2 internal nodes.
#>
#> Tip labels:
#> 3, 4, 5
#> Node labels:
#> 1, 2
#>
#> Rooted; no branch length.
plot(ans)